How to Train your Genomics Models
/First open resource hosts trained machine-learning genomics models to facilitates their use and exchange

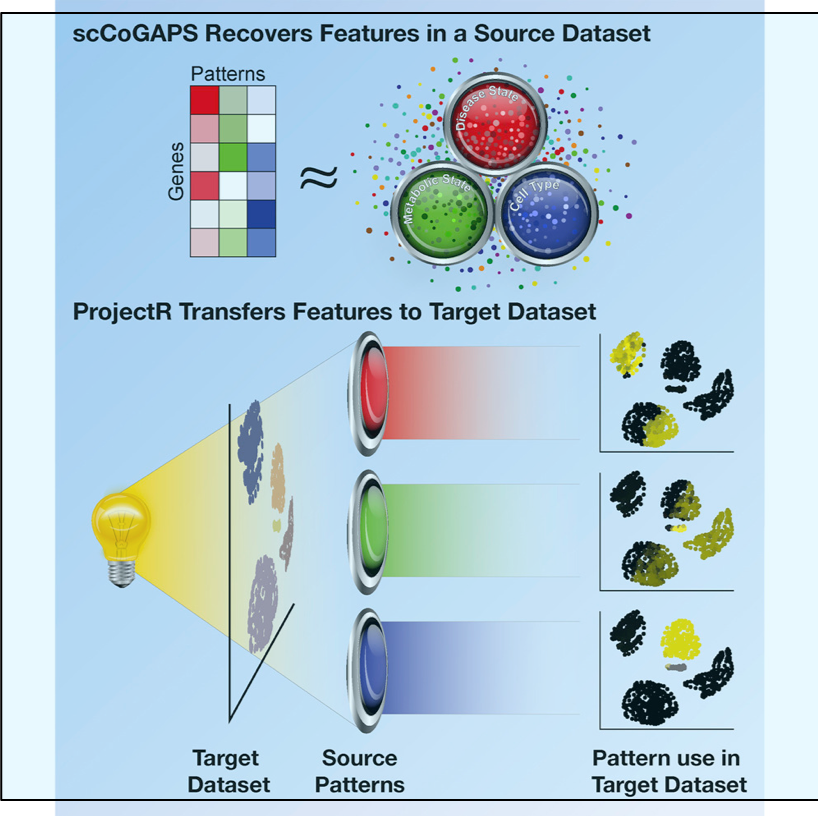
A powerful new resource, one that is actually a new kind of resource, has come online and, hopefully, will help accelerate advances in genomics and the fight against many types of disease. The scale of genome data is so large that computational tools are required for every major step of acquiring, organizing, and analyzing genomes. Generating useful models from large genomic datasets, the kind you generate when studying human disease, is often difficult and time consuming and many aspects of this are now being automated using various types of machine learning approaches. Machine learning in this context can be roughly summarized as using computers to generate and evaluate huge numbers of statistical models in order to clarify relationships in datasets. To do this, the machine learning program needs to train on useful datasets. So for many cutting edge applications, the program doesn’t just need to be written but also trained—and this second step can require large amounts of time and computational resources, making the transmission and broader application of these programs less likely, until now. The Kipoi repository is the first open resource for machine learning methods in genomics, making cutting edge approaches available to clinicians and smaller labs. This resource is sure to speed the application and innovation in machine learning based genomics approaches, and hopefully we will all benefit from this new site for the free exchange of ideas.
For more information, here’s a nice summary from Technology Networks.
Here is the introduction from the original article, published in Nature Biotechnology.
Advances in machine learning, coupled with rapidly growing genome sequencing and molecular profiling datasets, are catalyzing progress in genomics1. In particular, predictive machine learning models, which are mathematical functions trained to map input data to output values, have found widespread usage. Prominent examples include calling variants from whole-genome sequencing data2,3, estimating CRISPR guide activity4,5 and predicting molecular phenotypes, including transcription factor binding, chromatin accessibility and splicing efficiency, from DNA sequence1,6,7,8,9,10,11. Once trained, these models can be probed in silico to infer quantitative relationships between diverse genomic data modalities, enabling several key applications such as the interpretation of functional genetic variants and rational design of synthetic genes.
However, despite the pivotal importance of predictive models in genomics, it is surprisingly difficult to share and exchange models effectively. In particular, there is no established standard for depositing and sharing trained models. This lack is in stark contrast to bioinformatics software and workflows, which are commonly shared through general-purpose software platforms such as the highly successful Bioconductor project12. Similarly, there exist platforms to share genomic raw data, including Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and the European Nucleotide Archive (https://www.ebi.ac.uk/ena). In contrast, trained genomics models are made available via scattered channels, including code repositories, supplementary material of articles and author-maintained web pages. The lack of a standardized framework for sharing trained models in genomics hampers not only the effective use of these models—and in particular their application to new data—but also the use of existing models as building blocks to solve more complex tasks.