Excitement over the Single-Cell Genomics Market
/Single-Cell Genomics Market Expected To Reach A Highest Growth During Forecast Period

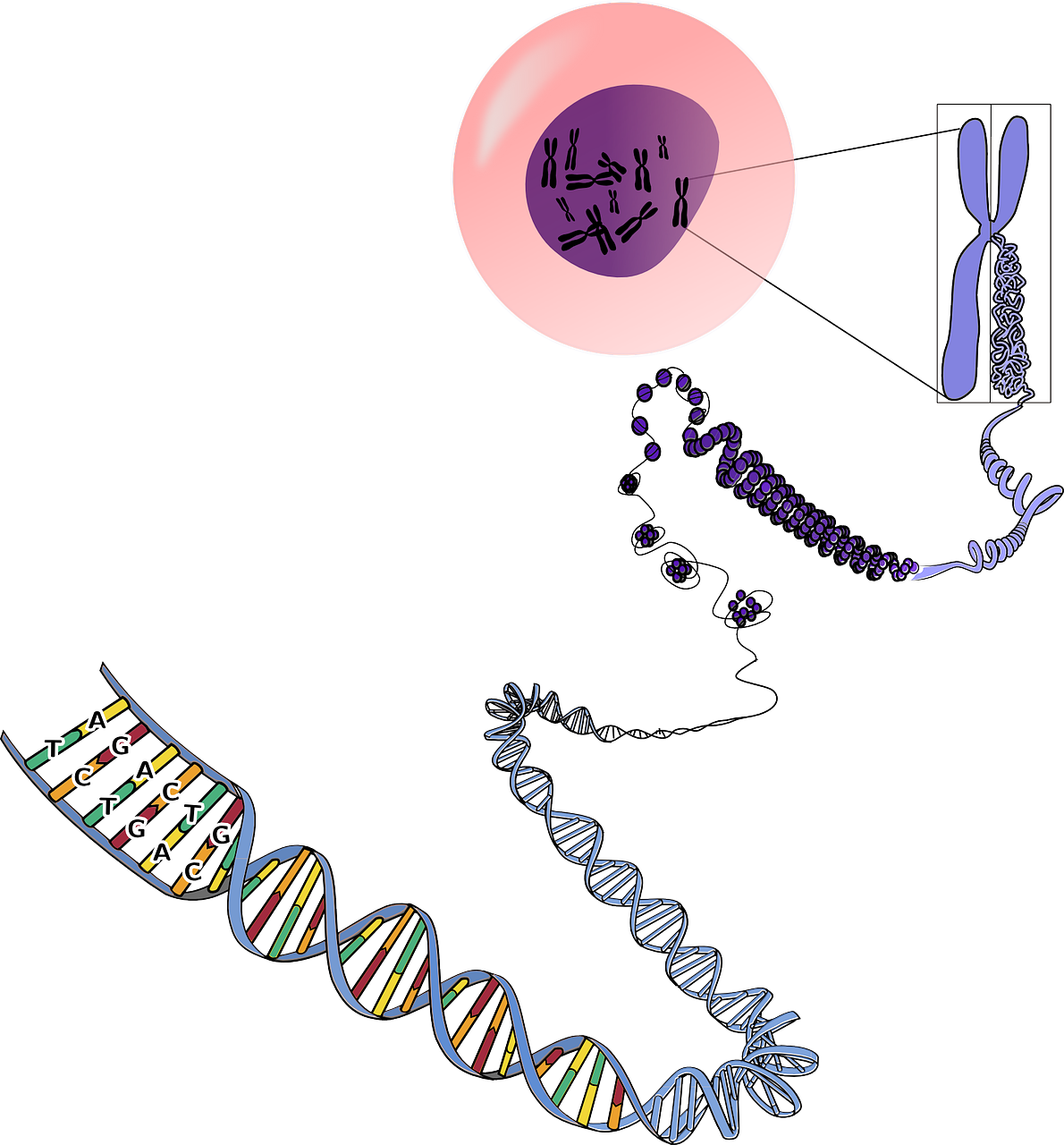
Single Cell Genomics is a rapidly growing market due to the new emerging methodologies in which the genomic technologies are applied at the single cell level, rather to all the cells collectively. The single cell genomic technologies are opening new boundaries by separating the contributions of single cells to the diversity of ecosystem and organisms. The single cell genomics is also creating new insight into multifaceted biological systems that range from the microbial ecosystem diversity to the human cancer genomics. To mention an example, the single cell genomics can probably be used to identify as well as assemble the genomes of the microorganisms which cannot be cultured, single cell genomics also evaluates the part genetic mosaic plays in the normal physiology and also determines the role of intra tumor genetic variation responsible for cancer development or treatment. However, the single cell genomics has the ability to evaluate a single DNA molecule from single isolated cells, but the process is technically challenging.